INTRODUCTION
Fabry disease is a rare, underdiagnosed X-linked metabolic condition with potentially fatal systemic consequences. Fabry disease is characterized by two recognized phenotypes: the classic, early-onset type is found in the pediatric population and associated with neuropathic pain, whereas the later-onset type is predominantly associated with cardiac manifestations.1 Fabry disease often presents with unique ocular manifestations identifiable during a routine eye examination. Clinical ocular findings most commonly include corneal opacities (referred to as cornea verticillata for the remainder of this case report) and less commonly as conjunctival vascular abnormalities, lens opacities, and retinal vascular abnormalities.1 Eye care providers can play a critical role in helping patients with undiagnosed Fabry disease by recognizing the cornea verticillata that is pathognomonic for Fabry disease and referring patients with ocular findings for genetic testing.1–3
This case describes a patient previously undiagnosed with Fabry disease who presented with classical ocular and systemic signs and symptoms and introduces vitreous hemorrhage as a presenting ocular sign of Fabry disease. A general overview of the disease with emphasis on the importance of early detection of Fabry disease for optimal disease management is also discussed.
CASE REPORT
Initial Visit
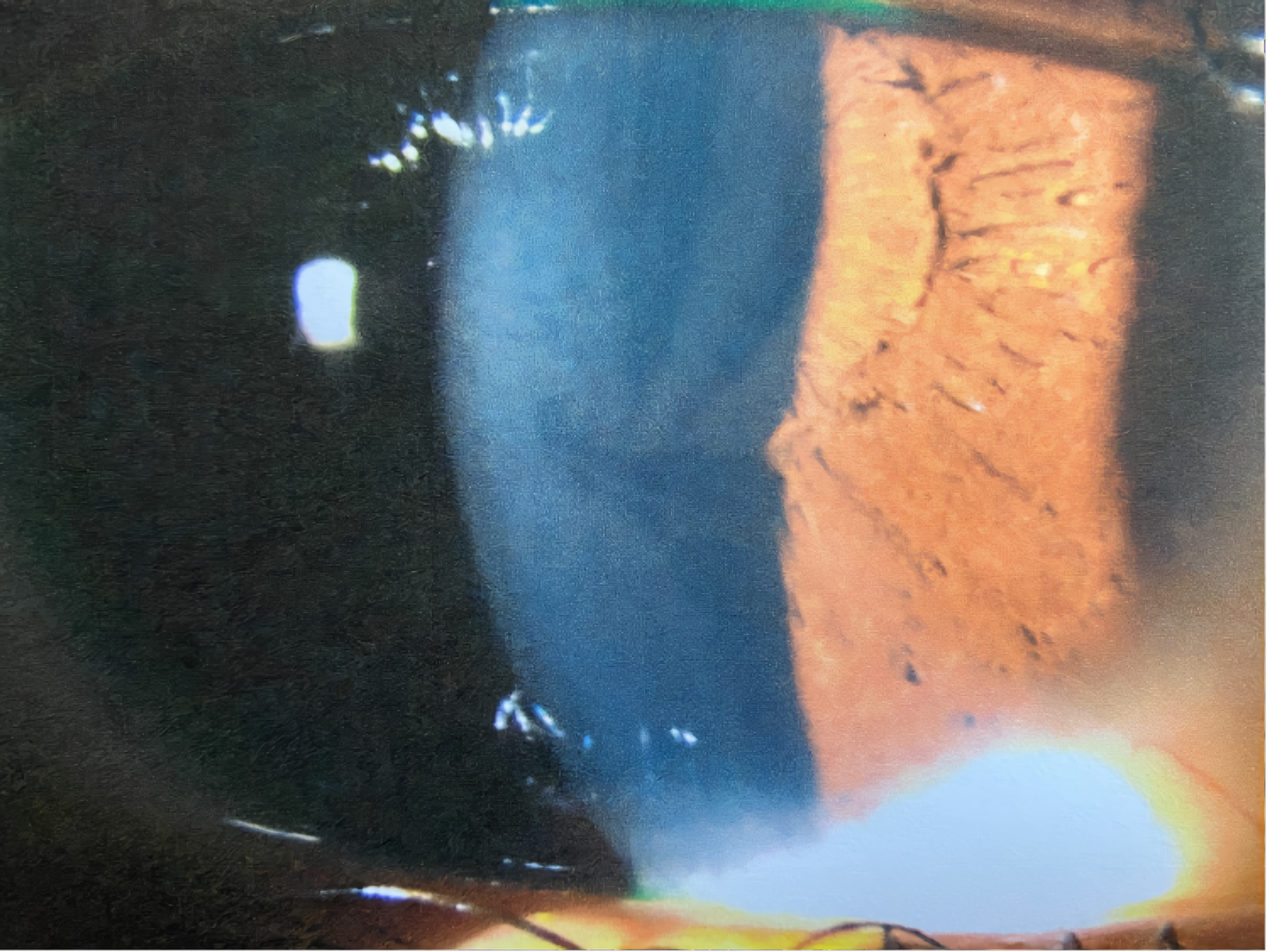
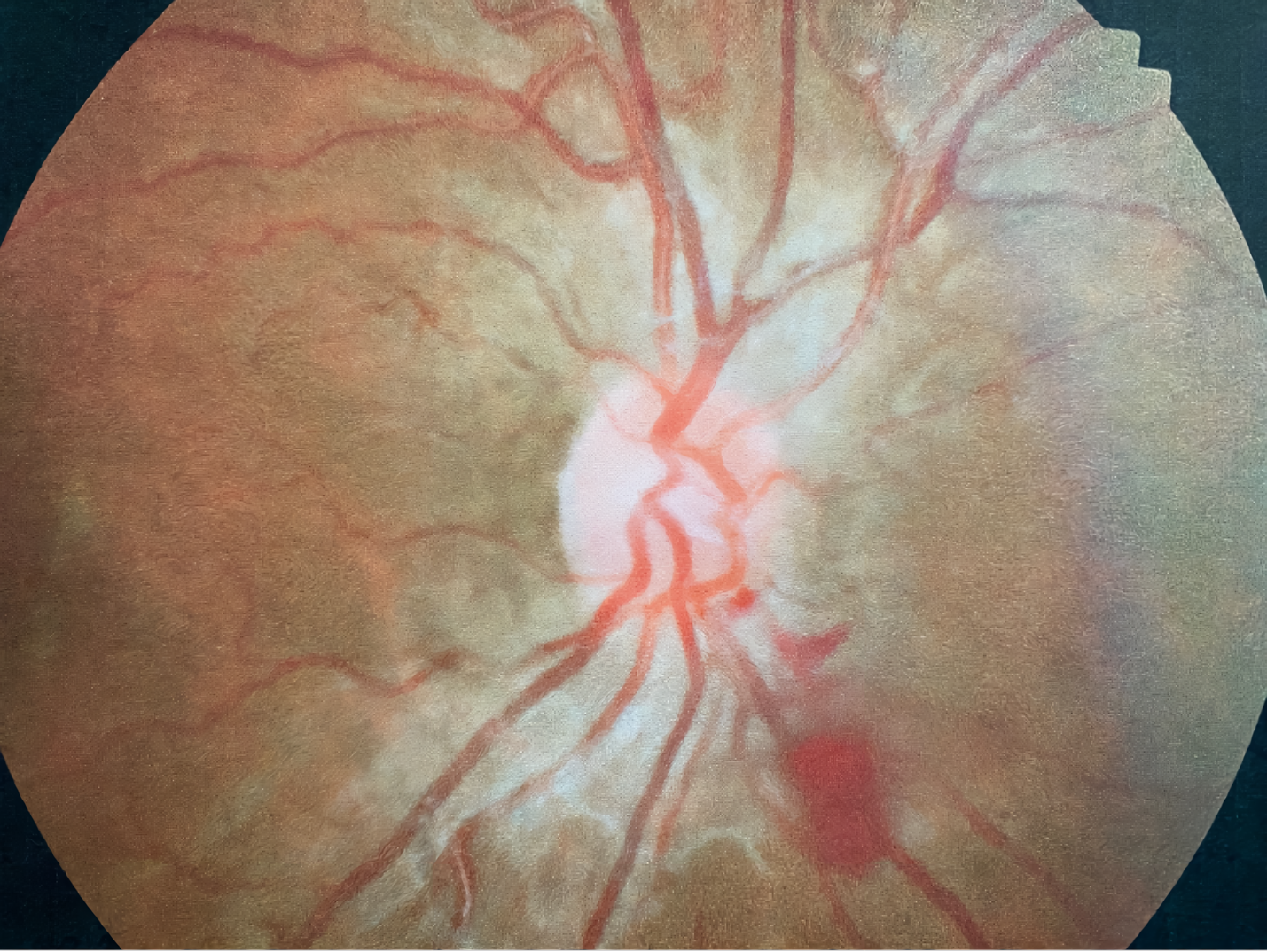
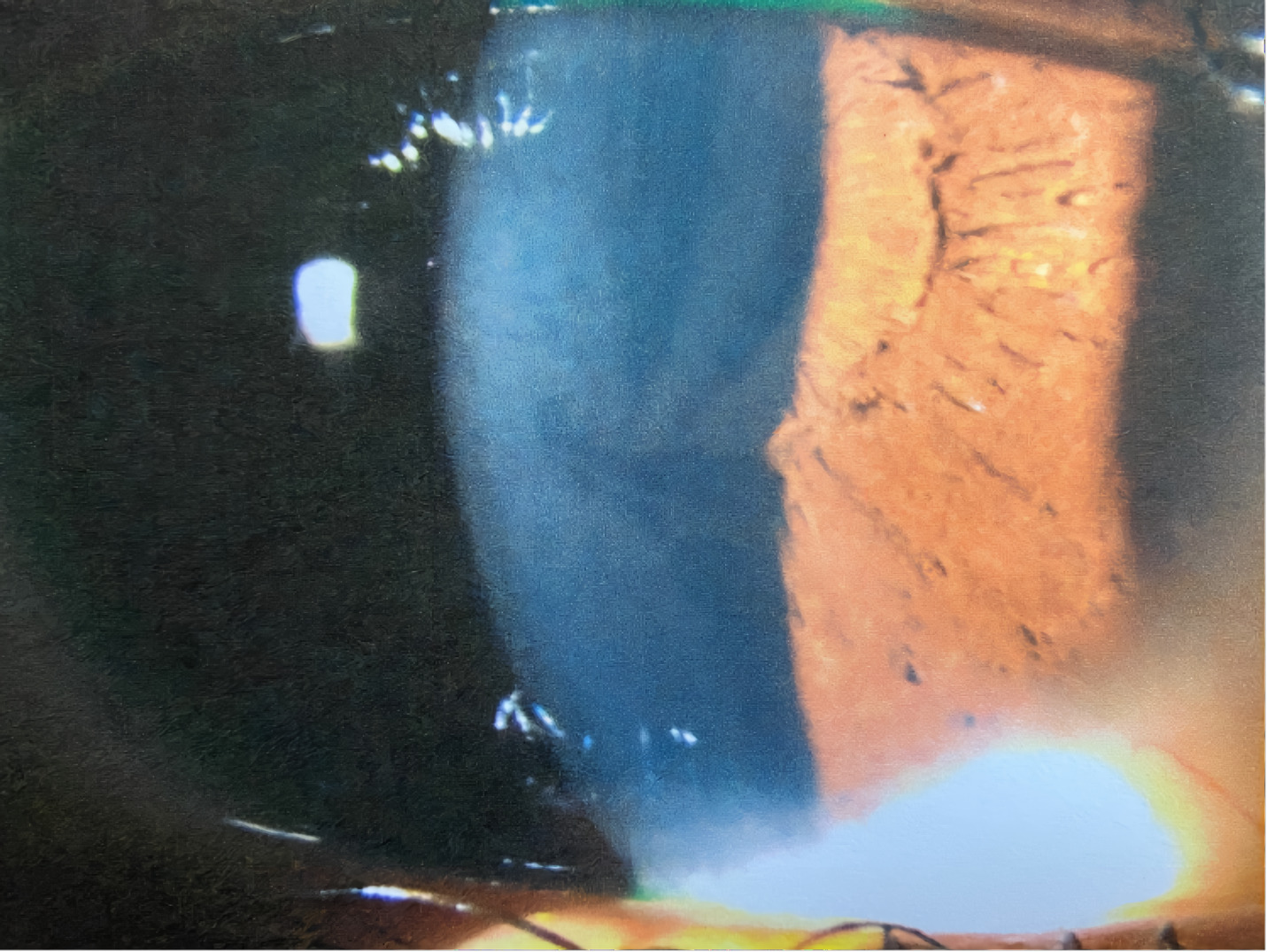
A 21-year-old African American woman presented at Navy bootcamp with complaints of a sudden-onset black spot in her right eye that occurred immediately after vomiting earlier that day. Aside from a complaint of acute gastrointestinal pain, the patient’s medical history was unremarkable. Visual acuity was 20/20 in each eye. Confrontation visual fields were full to finger counting in both eyes. Extraocular muscle and pupil testing were unremarkable. Slit lamp biomicroscopy was remarkable for diffuse brownish corneal epithelial deposits with a central whorl-like pattern in both eyes (Figure 1). Intraocular pressures were right eye 13 mm Hg and left eye 12 mm Hg by noncontact tonometry. Dilated fundus evaluation was remarkable for vessel tortuosity in both eyes and a small vitreous hemorrhage inferior-nasal to the optic disc in the right eye (Figure 2). Posterior segment findings of the optic nerve, macula, and periphery were otherwise normal in both eyes.

Blood pressure measured 118/72. The patient denied floaters, flashes of light, or curtain or veil in her vision. She also denied history of ocular trauma or surgery to either eye, and she had no known history of any systemic conditions. Further questioning exposed a strong family history of cardiovascular disease, cerebrovascular disease, and kidney failure. The patient’s gastrointestinal pain had waxed and waned over the previous few weeks, but this was the first instance of vomiting or associated ocular symptoms.
The patient was diagnosed with bilateral whorl keratopathy and Valsalva retinopathy in the right eye. Given the clinical presentation and genetic predisposition with gastrointestinal pain and history of vomiting, she was referred for blood work specific for Fabry disease. The patient was educated on the self-resolving nature of the vitreous hemorrhage and that vessel tortuosity and vomiting put her at increased risk for its occurrence. No retina referral was recommended at this visit, but the patient was told to avoid blood thinners and to return in 1 week for a follow-up visit.
Differential Diagnoses of Superficial Corneal Deposits
-
Cornea Verticillata: whorl-like brown or white opacities within the epithelial basement membrane. This is the primary differential diagnosis based on the examination findings.
-
Amiodarone Keratopathy: amiodarone, an anti-arrhythmic agent used to treat various cardiac dysrhythmias, causes whorl keratopathy in 70%-100% of patients.4 Chloroquine, hydroxychloroquine, indomethacin, and tamoxifen use can result in a similar presentation of whorl keratopathy. This patient had no history of amiodarone or other medication use.
-
Hudson-Stahli Line: an iron deposition line in the corneal epithelium and typically seen inferiorly in a linear pattern. The clinical presentation in this patient was more extensive than a single linear or ring-like pigment deposit, ruling out Hudson-Stahli as a diagnosis.
-
Spheroidal Degeneration: a degeneration of the cornea and/or conjunctiva characterized by bilateral, translucent spherules in the cornea. Our patient presented with a corneal pattern inconsistent with translucent spherules.
Differential Diagnoses for Vitreous Hemorrhage
-
Valsalva Retinopathy: vomiting or coughing resulting in preretinal hemorrhaging.5 A positive and recent history of vomiting with no known systemic conditions makes this the lead differential diagnosis in this patient.
-
Terson Syndrome: associated with sudden elevated intracranial pressure, Terson syndrome hemorrhaging can present in the subhyaloid space or subinternal limiting membrane, both of which are considered vitreous hemorrhages. The patient did not suffer from elevated intracranial pressure, so this condition was ruled out.
-
Retinal Macroaneurysm: focal dilations of retinal arterial branches. Bleeding can be subretinal, intraretinal, sub–internal limiting membrane, or within the subhyaloid space. Patients with retinal macroaneurysm typically have hypertension; this patient had normal blood pressure in-office.
Differential Diagnoses for Retinal Vessel Tortuosity
-
Hypertensive Retinopathy: a retinal disorder that can result in scattered flame-shaped hemorrhages within the posterior pole, specifically within the retinal arcades, and notable retinal vessel tortuosity. The patient did not have hypertension.
-
Retinal Vein Occlusion: another retinal disorder in which retinal vessel tortuosity with extensive intraretinal hemorrhaging is observed throughout the posterior pole. Although this patient had retinal vessel tortuosity, the isolated heme is not consistent with retinal vein occlusion.
Follow-up Visit #1
At the 1-week follow-up appointment, the patient reported an improvement in symptoms and that the spot had faded in the right eye. Visual acuity was stable in both eyes, as was entrance testing. Anterior segment findings and intraocular pressures were stable in both eyes. Because the patient was a Navy recruit in a high-volume in-processing clinic, repeat dilation was not performed. The vitreous hemorrhage, as observed with undilated posterior segment evaluation, was noted to be resolving due to its smaller size.
The patient was reeducated on the self-resolving nature of the vitreous hemorrhage given its improvement both objectively and subjectively. She was asked to return to the clinic in 1 week.
Follow-up Visit #2
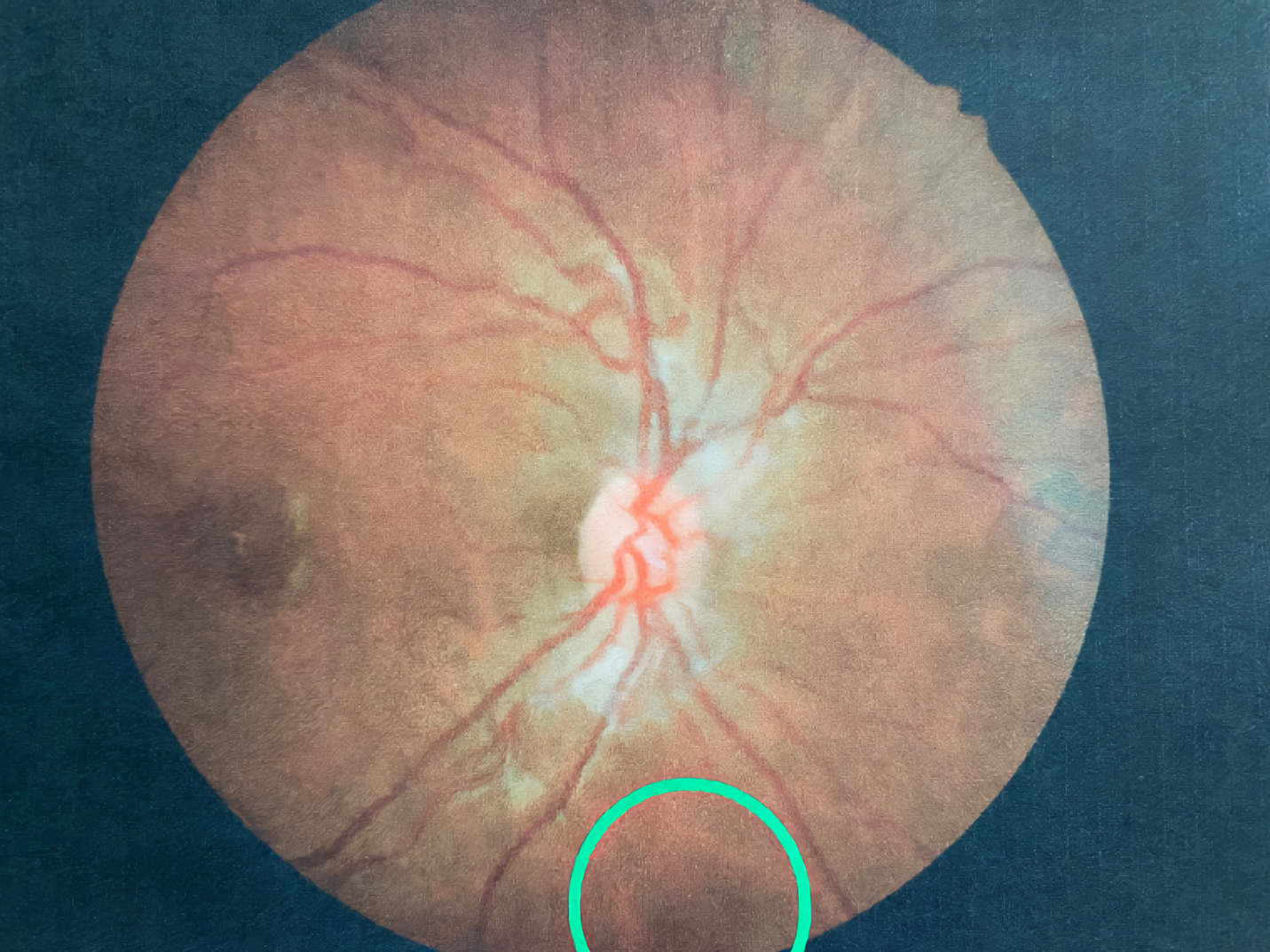
At the 2-week follow-up appointment, the patient reported that the spot was gone and denied any other ocular or visual complaints. Vision and slit lamp findings were stable in both eyes. Undilated fundus evaluation was remarkable for mild vessel tortuosity in both eyes and complete resolution of the vitreous hemorrhage in the right eye (Figure 3).

Blood plasma testing confirmed a below-normal level of α-galactosidase A consistent with heterozygous Fabry disease. Upon receiving the confirmatory laboratory results, the patient was educated about the disease and treatment options, including enzyme replacement therapy. She was medically discharged from the Navy, referred to her primary care physician, and advised to return annually for eye examinations or sooner if any changes in vision were noted.
DISCUSSION
Fabry disease is a rare, X-linked lysosomal storage disorder that affects anywhere from 1:40,000 to 1:117,000 individuals; these are likely underestimations owing to missed or overlooked clinical signs and symptoms.4,6,7 Presentation of classical Fabry disease includes neuropathy, gastrointestinal disturbances (abdominal pain), hypohidrosis (sweating abnormalities), and angiokeratomas (skin abnormalities).7–9 Symptoms typically begin in adolescence (approximately age 10) and can lead to severe multiorgan dysfunction and premature death if not detected. The disease stems from deficiency of the enzyme α-galactosidase A that leads to an accumulation of glycophospholipids. α-galactosidase A catalyzes a breakdown of globotriaosylceramide to galactosylceramide, the lack of which results in accumulation of globotriaosylceramide in lysosomes and triggers a cascade of inflammation in affected tissues.7 Fabry disease is diagnosed with confirmed low α-galactosidase A levels via fluorometry, a method for detecting fluorescence that uses ultraviolet light to stimulate the enzyme.7
Fabry disease affects male patients at a younger age and more severely compared with female patients.2 Homozygous male patients present with the classical variant, whereas heterozygous female patients have a wider spectrum of disease presentation, likely because of lyonization, where one X chromosome is inactivated.1 “Classic Fabry disease” was initially described in male patients with a severe clinical phenotype due to either the absence or significant reduction of α-galactosidase A activity. It was associated with childhood onset and progressive, irreversible multiorgan failure.7,8 Heterozygous female patients may find that clinical manifestations appear 10 years later on average than those in male patients.9
Ocular findings in Fabry disease include eyelid swelling, conjunctival vessel varicosities, cornea verticillata, cataract, central retinal artery occlusion, visual field defects, internuclear paralysis of extraocular muscles, and occasional edema of the optic disc and retina. Cornea verticillata, cataract, and vascular tortuosity of the conjunctiva and retina have been most documented with Fabry disease10 and have a high specificity for Fabry disease.9
Corneal verticillata affects the corneal epithelium and basement membrane, excluding stromal and endothelial involvement.2 This finding is the most common ocular manifestation of Fabry disease, affecting up to 88% of female and 95% of male patients.3,10 It often occurs bilaterally, symmetrically, and with a vortex pattern of pigmented subepithelial or intraepithelial globotriaosylceramide deposits that are taken up by the lysosomes of limbal epithelial cells that differentiate and migrate centrally, forming a whorl pattern.4 A fundamental optical property of the cornea is transparency; however, verticillata neither affects visual acuity nor causes visual symptoms.2
There are conflicting data on the correlation of cornea verticillata to disease severity as well as changes to cornea verticillata over time. Pitz et al. identified cornea verticillata as a predictor for severity of Fabry disease. Using the Fabry Outcome Survey database, a correlation was made between prevalence of ocular change in patients with Fabry disease and disease severity. Their analysis suggests that patients with Fabry disease with ocular findings have higher disease severity than those without ocular findings.11 Conversely, Moiseev et al. found that although there was a strong association of Fabry disease with ocular presentation of cornea verticillata, there was no association between cornea verticillata and severity of Fabry disease.12 A retrospective cross-sectional study by Mete et al. also found no association between cornea verticillata and disease severity.12
Sivley and Benjamin investigated whether cornea verticillata can worsen over time or improve with treatment. All untreated patients had observable changes of keratopathies, whereas stability or improvement was noted in patients undergoing treatment for the duration of the study.13 Although corneal involvement is variable over time among these studies, cornea verticillata does have a strong general association with Fabry disease. Samiy created a table of questions to ask a patient with cornea verticillata (Table 1) to aid in diagnosis determination. These questions help strengthen the suspicion for Fabry disease if answered positively and can influence whether to order laboratory work that ultimately confirms a diagnosis of Fabry disease.
Other ocular findings of Fabry disease include less visible conjunctival vascular abnormalities, cataracts, and retinal vessel tortuosity. Although the isolated presence of tortuous vessels without lens or corneal findings is not specific to Fabry disease, their presence with corneal findings may indicate more significant disease severity. Posterior subcapsular opacities associated with Fabry disease, known as “Fabry cataracts,” are more rare but also more specific for Fabry disease. Retinal vascular tortuosity occurs secondary to substrate accumulation in the vascular endothelium.10
Unique to this case is the presentation of vitreous hemorrhage. Vitreous hemorrhages are supplied by the central retinal artery and present within the vitreous anteriorly. The mechanisms for the pathogenesis of vitreous hemorrhage can include retinal vascular disorders with or without associated ischemia, breakthrough vitreous hemorrhage, or rupture of blood vessels (i.e., neovascularization).14 Blood in the vitreous occurs as an inflammatory reaction formed from polymorphonuclear neutrophils that break down fibrin macrophages then phagocytose red blood cells and cellular debris.15 Increased intra-abdominal pressure associated with an absence of valves in the venous system anterior to the heart leads to an increase in intraocular pressure and subsequent rupture of superficial retinal capillaries.12 The presentation of a vitreous hemorrhage in this case was likely due to the patient’s gastrointestinal distress, which is consistent with Fabry disease, and subsequent vomiting/Valsalva maneuver.
There are no documented cases of Fabry disease with Valsalva retinopathy. The Valsalva maneuver, otherwise known as hemorrhagic retinopathy of Valsalva, is the rupture of superficial capillaries secondary to an increase in retinal venous pressure following a sudden change in intrathoracic or intra-abdominal pressure.12 A rapid rise of intraocular venous pressure can cause spontaneous rupture of the superficial retinal capillaries, potentially leading to sudden and painless decrease in visual acuity in an otherwise healthy eye.14 Vessel tortuosity may have also put this patient at increased risk for hemorrhaging owing to weaker vasculature. Depending on the size of the vessel affected, Valsalva retinopathy can vary in presentation. It is largely self-limiting in nature but can require surgical intervention if larger or if resolution is not seen after 3 weeks of observation. In this case, gastrointestinal distress associated with Fabry disease was the presumed cause of this episode of vomiting, and the hemorrhage resolved without treatment.
The diagnosis of Fabry disease is largely based on clinical signs and symptoms. Organ involvement can be diagnostic, and impaired kidney function tests or echocardiography can confirm the diagnosis.8 Laboratory testing for α-galactosidase A deficiency should be performed in cases of suspected Fabry disease. Molecular genetic testing can also be performed and confirmed in the presence of the galactosidase α gene. Upon diagnosis, patients with Fabry disease should receive genetic counseling and family screening. These interventions are helpful in identifying additional familial cases, provide the patient with a better understanding of their condition, and identify the risk of offspring inheriting the disease.8 Where applicable, psychosocial, economic, insurance, and family planning considerations should be discussed with patients in their counseling sessions.
Although analgesics or carbamazepine can be prescribed for pain management, treatment of Fabry disease is focused on replacing the absent or deficient α-galactosidase A enzyme by means of enzyme replacement therapy.8,16 Introduced in 2001, enzyme replacement therapy was approved by the United States Food and Drug Administration in 2003. There are two forms of recombinant α-galactosidase A: Agalsidase α (Replagal) is produced by continuous human cell lines; Agalsidase β (Fabrazyme) is a recombinant form of the deficient enzyme produced by Chinese hamster ovary cells transduced with the α-galactosidase A gene. The United States Food and Drug and Administration has approved Fabrazyme for Fabry disease. The recommended dosage is 1 mg/kg of body weight, administered every 2 weeks as an intravenous infusion.1,16 In 2014, another agalsidase β, Fabagal, was approved for use in South Korea.17
Enzyme replacement therapy is considered the standard of care for patients with Fabry disease and has been found to have a positive impact on both systemic and ocular complications. Enzyme replacement therapy studies show promising results; improvements in pain level, cardiovascular function, and renal function have all been noted.16 A study by Germain et al. showed the predictive importance of younger age and absence of organ damage when enzyme replacement therapy is initiated.18 Fledelius et al. found that 41% of patients who were observed over 10 years following initiation of enzyme replacement therapy showed a reduction of cornea verticillata without an effect on visual acuity over the study period.19 When treated with enzyme replacement therapy for 10 years, patients showed stabilization or slowing progression of the disease.16 No studies have provided a definitive recommendation for the duration of enzyme replacement therapy, but because the drug is rapidly depleted in the body, treatment is believed to be lifelong.8
Due to the cost and complexity of intravenous infusion delivery treatment, other therapies are being investigated. Chaperone drugs can aid in patients with unstable variants of the mutant α-galactosidase A enzyme. Migalastat (Galafold) is the only oral chaperone drug Federal Drug Administration approved for Fabry disease treatment. The drug has shown an effect comparable to that of enzyme replacement therapy on renal function and cardiac outcomes in a clinical trial, but only for eligible patients with specific mutations.17 A small molecule, the chaperone does not induce antidrug antibodies like enzyme replacement therapy does, making it more effective at crossing the blood-brain barrier.
Plant-derived enzyme replacement therapies are designed to enhance efficacy by increasing plasma half-life and reducing immunogenicity.16 Substrate reduction therapy is an alternative means of enhancing enzyme efficacy. Substrate reduction therapy is a small molecule iminosugar that carries similar benefits to chaperone therapy like Galafold; it has shown potential in patients with Fabry disease.16 Gene therapy aims to add a normal α-galactosidase gene to the patient’s DNA, theoretically inducing the ability to produce normal enzyme. Systemic messenger RNA therapy carries some advantages over enzyme replacement therapy but also requires repeated administration to remain effective. Last, heart transplantation followed by enzyme replacement therapy is a potential treatment option for select patients but presents its own risks and is prone to a Fabry disease effect on the donor graft.16
Treatment with enzyme replacement therapy should be combined with supportive management to address each patient’s systemic symptoms and complications. Preventive measures such as stroke prophylaxis and lifestyle modifications (smoking cessation, dietary salt restriction, and treatment of hypertension and dyslipidemia) are also important recommendations. Last, periodic comprehensive ocular health examinations are an important part of thorough management.20
CONCLUSION
This case represents a unique opportunity for eye care practitioners to play a vital role in the early detection of a potentially fatal genetic condition. Vitreous hemorrhage presenting as Valsalva retinopathy in the presence of more classic ocular signs of Fabry disease has not been previously noted in the literature but should be considered a potential presenting sign of the condition. Identifying such signs and symptoms should raise suspicion of Fabry disease and prompt a referral for the appropriate testing and treatment.
Conflicts of Interest
No identifiable health information was included in this case report. The author declares no conflict of interest.
TAKE HOME POINTS
-
Fabry disease is a potentially fatal condition that presents with unique ocular findings that can be identified with a routine eye examination.
-
Whorl keratopathy (cornea verticillata) in the absence of other systemic conditions or medication use is highly suggestive of Fabry disease.
-
Knowing the systemic manifestations of Fabry disease, such as gastrointestinal distress leading to valsalva retinopathy, can also point in the direction of appropriate lab work for confirmation of the disease.
ACKNOWLEDGMENTS
Many thanks to David Malchow, OD, who took part in this patient’s care. Thank you also to Raman Bhakhri, OD, for his mentorship.