

INTRODUCTION
Horner syndrome is a rare condition caused by disruption of the sympathetic nerve pathway to the eye. It is characterized by a classic triad of symptoms including ptosis, miosis, and anhidrosis, although iris heterochromia, inverse ptosis of the lower eyelid, and pupillary dilation lag may also be present. It can be classified into congenital or acquired types. The congenital form is most commonly caused by birth trauma, and the acquired form is usually due to damage to the sympathetic nerve pathway by surgical intervention or trauma but can also be caused by tumors, infections, or vascular malformations.1
Neuroblastic tumors are the most common extracranial solid tumors in infants, particularly during the first 2 years of life. In the United States, the incidence is approximately 1 in 7000 live births.2 The majority of neuroblastomas occur sporadically, although some rare familial cases do exist. Neuroblastomas are the most common occult malignancy linked to pediatric Horner syndrome. However, research indicates that only 3.5% to 13% of children with neuroblastoma have associated Horner syndrome, whereas in only 2.2% of cases, Horner syndrome is the initial symptom.3,4
This report describes a 2-year-old girl presenting with acquired Horner syndrome secondary to mediastinal ganglioneuroblastoma. No identifiable health information was included in this case report.

CASE REPORT
A 2-year-old female patient presented with a complaint of unequal pupil sizes with the right pupil being larger than the left since 10 months of age. The patient’s parents noted that the pupils may be closer in size in the dark and that the patient’s irises also may be slightly different in color. They did not report one-sided sweating or facial flushing. The patient’s parents reported no significant birth, medical, or surgical history, no developmental concerns, no recent head or ocular trauma, and no visual or ocular symptoms.
On examination, the uncorrected visual acuity was 20/30 in both eyes tested with forced-choice isolated Lea optotypes with crowding bars. Color vision screening with F2 plates and contrast sensitivity testing with Mr. Happy was normal. Stereoacuity was measured as 60 seconds of arc with the Pediatric Assessment of Stereopsis with a Smile Test (Vision Assessment Corporation). The patient was orthophoric at distance and near with no restrictions in ocular motility of either eye. The right pupil was measured to be 6 mm in dim lighting and 4.5 mm in bright lighting, whereas the left pupil was measured to be 5 mm in dim lighting and 3.5 mm in bright lighting. Pupils were round and equally reactive to light, and no relative afferent pupillary defect was noted. Anterior segment evaluation revealed slight heterochromia, and the right iris appeared slightly more pigmented than the left, with mild lid asymmetry (Figure 1). Palpebral aperture size was measured to be 10 mm in both eyes. Cycloplegic retinoscopy revealed a low hyperopic and astigmatic prescription that did not require correction, and fundus examination was unremarkable in both eyes.

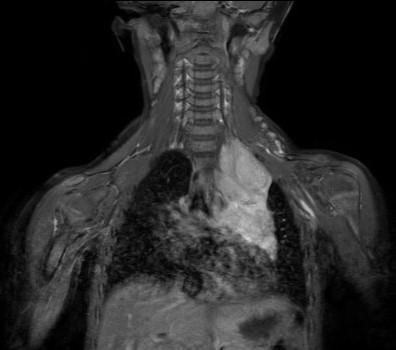
Differential diagnoses that could be considered for congenital iris heterochromia include Waardenburg syndrome and Sturge-Weber syndrome. These were excluded due to the lack of hearing loss, pigmentation abnormalities, and facial port-wine stains. Given the pupil asymmetry, iris heterochromia, and suspicion of Horner syndrome, the patient was referred to pediatric ophthalmology for further evaluation including magnetic resonance imaging. At the follow-up 3 days later with the ophthalmologist, the patient’s right pupil was measured to be 5 mm in dim lighting and 3.5 mm in bright lighting, whereas the left pupil was measured to be 3.5 mm in dim lighting and 2.5 mm in bright lighting. It was also noted that the upper lid crease of the left eye appeared fuller than that of the right eye. Magnetic resonance imaging of the head, neck, and upper thorax was recommended, which led to the discovery of a mediastinal ganglioneuroblastoma (Figure 2), resulting in a second-order Horner syndrome.

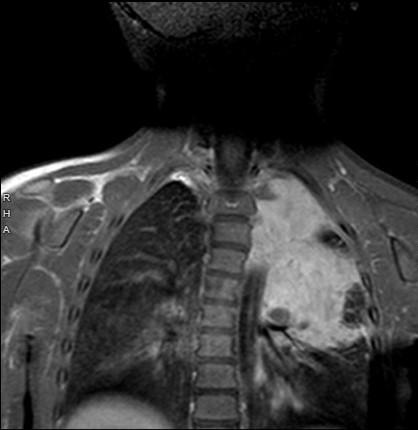
A follow-up examination of the patient 1 year after partial resection of the ganglioneuroblastoma (Figure 3) revealed persistent paralytic left ptosis, with the right eye’s marginal reflex distance measured at 5 mm and the left eye at 3.5 mm. The anisocoria remained stable, with the patient’s right pupil measuring 5 mm in dim lighting and 3.5 mm in bright lighting, whereas the left pupil measured 3.5 mm in dim lighting and 2.5 mm in bright lighting. The patient maintained equal visual acuity in both eyes, so it was recommended to continue monitoring the condition without additional intervention.

DISCUSSION
Horner syndrome is a rare neurological condition that results from disruption of the sympathetic nerve pathway and classically presents with ptosis, miosis, and anhidrosis. In the pediatric population, it can be classified as congenital (55% of cases) or acquired (45%).1 The sympathetic pathway involved in Horner syndrome includes 3 orders of neurons. The first-order neurons travel from the hypothalamus, through the brainstem, and down the spinal cord to the lower cervical and upper thoracic regions. The second-order neurons exit the spinal cord, ascend through the chest cavity over the apex of the lung, and reach the superior cervical ganglion in the neck. Third-order neurons leave the superior cervical ganglion and follow the internal carotid artery through the cavernous sinus to the eye or along the external carotid to the face.5 Damage to any of the 3 neurons along this pathway, such as from a stroke, tumor, or trauma, can lead to the development of Horner syndrome with all symptoms occurring on the same side as the lesion. In pediatric cases, Horner syndrome is often associated with a lesion affecting the second-order neuron as seen in this case, with the tumor location in the left lung apex.
The ptosis in Horner syndrome is caused by dysfunction of Müller’s muscle, usually leading to a mild ptosis of 1 to 2 mm in the upper eyelid. Although less common, the same sympathetic dysfunction can affect the inferior tarsal muscle, causing an inverse ptosis of the lower eyelid. Paresis of the dilator pupillae muscle causes anisocoria, resulting in a smaller but still reactive pupil on the affected side. A subtle dilation lag, in which the affected pupil dilates more slowly or does not dilate as fully as the unaffected pupil, may also be noticeable in the dark. Anhidrosis occurs due to the disruption of vasomotor and sudomotor nerve innervation. The extent of anhidrosis can vary depending on the location of the lesion, ranging from affecting an entire half of the face to a small patch on the forehead.5 In some cases, Harlequin sign, or hemifacial flushing, may occur as a compensatory response on the unaffected side to the sympathetic dysfunction on the affected side.6 It is typically triggered by exercise, emotional stress, or heat and can be more apparent than anhidrosis. Iris heterochromia is commonly seen in congenital Horner syndrome but can also develop in cases of Horner syndrome acquired before the age of 2 as seen in this case. Lack of sympathetic stimulation to the iris melanocytes, which affects melanin production, results in a lighter-colored iris on the affected side.
Diagnosing Horner syndrome in children can be challenging because this population can be difficult to examine and the ptosis and miosis may be subtle, occur intermittently, or not appear simultaneously.7,8 Intermittent symptoms may be an early indication of Horner syndrome developing as a result of variable compression of the sympathetic chain. This may have contributed to the variation in examination findings observed between the initial visit and the ophthalmology follow-up just 3 days later.
Pharmacological testing with cocaine has traditionally been used to confirm the diagnosis of Horner syndrome as the normal pupil dilates more than the affected pupil. Apraclonidine, which dilates the affected pupil more than the normal pupil, can be used as an alternative but has safety concerns in the pediatric population.5 Hydroxyamphetamine can help differentiate between preganglionic and postganglionic lesions, but it has been shown to be less reliable in younger patients.9 Both cocaine and hydroxyamphetamine are no longer commercially available. Given these concerns, the current recommendation is to perform imaging in the absence of significant birth history.5
Neuroblastoma is the most common occult malignancy linked to pediatric Horner syndrome. This type of cancer originates from neuroblasts and typically develops in the adrenal glands or along the sympathetic nervous system, often in the abdomen or chest. When the tumor affects the sympathetic nerve fibers, it can lead to Horner syndrome, especially in infants and young children. Approximately 95% of neuroblastomas and ganglioneuroblastomas occur in patients aged younger than 5 years.2 Most neuroblastomas occur sporadically with an unknown cause. The cellular characteristics and early onset of the disease suggest a role for genetic factors and perinatal exposure. Mediastinal neuroblastomas typically have a favorable prognosis, with a 5-year survival rate of 90% for both neuroblastoma and ganglioneuroblastoma.2 Favorable prognostic factors include a diagnosis before 18 months of age.
In cases of pediatric Horner syndrome, the neuroblastoma may be silent or “occult,” meaning the tumor might not present obvious symptoms apart from the signs of Horner syndrome. As a result, neuroblastoma should always be considered in the differential diagnosis when a child presents with Horner syndrome, even if there is no visible mass initially. Early detection through imaging techniques like ultrasound, computed tomography scans, or magnetic resonance imaging is crucial for identifying neuroblastoma and initiating treatment, as it can be aggressive if not addressed promptly.2,5 Magnetic resonance imaging is preferred due to the risks of radiation exposure in young children. However, computed tomography scans are also sensitive to mass lesions, and the risk is considered acceptable if magnetic resonance imaging is unavailable or if there are concerns about the potential neurotoxicity associated with general anesthesia. Along with imaging of the entire oculosympathetic pathway, spot urine testing for urine catecholamines can be useful. One study found elevated levels in half of the neuroblastoma cases that underwent testing.10 Although nonspecific, elevated urine catecholamines increase the urgency for imaging. In this case, elevated urine catecholamines were observed, consistent with the presence of a ganglioneuroblastoma (Table 1).
Even after addressing the underlying cause, the ocular manifestations of Horner syndrome often persist. Treatment options include blepharoplasty for visually significant ptosis and addressing any amblyopia. Blepharoplasty can also be considered later for cosmesis as well as colored contact lenses to address any iris heterochromia. In adults, topical alpha adrenergic agonists such as oxymetazoline hydrochloride ophthalmic solution 0.1% (UPNEEQ, RVL Pharmaceuticals) have been shown to improve eyelid elevation.11
CONCLUSION
Although rare, this case highlights the importance for eye care providers to consider acquired causes of Horner syndrome when there is no history of birth trauma. Horner syndrome can present subtly and intermittently in pediatric patients. Given that it may be the first sign of a neuroblastoma, early diagnosis is crucial for effective management and treatment.
TAKE HOME POINTS
-
Iris heterochromia can be seen in acquired Horner’s syndrome before the age of 2.
-
Neuroblastoma is the most common occult malignancy linked to pediatric Horner syndrome.
-
If Horner syndrome is suspected, prompt imaging is essential.
ACKNOWLEDGMENTS
The author would like to thank Tina Chou, MD, for the follow-up care of the patient.