


INTRODUCTION
Neuromyelitis optica spectrum disorder, also referred to as neuromyelitis optica, is an autoimmune, demyelinating disorder of the central nervous system that can present with optic neuritis and transverse myelitis. Clinical presentation may include unilateral or bilateral visual decline that is often severe and can include an afferent pupillary defect. Optic nerve appearance may show no signs of papillitis as the inflammation is frequently retrobulbar, but mild papillitis can occur. Additionally, the onset of transverse myelitis and optic neuritis in neuromyelitis optica occurs sequentially and has high potential for relapse though it can be monophasic.1 Earlier thoughts suggested that neuromyelitis optica was classified within the same spectrum as multiple sclerosis; however, neuromyelitis optica is now considered a separate, unique diagnosis. More recent information identifies the presence of the serum autoantibody marker, aquaporin-4 immunoglobulin G, that is unique to neuromyelitis optica and not found in multiple sclerosis. As the name suggests, aquaporin-4 immunoglobulin G targets astrocytic protein aquaporin-4 of the blood-brain barrier within the central nervous system.2,3 Although prior criteria selectively emphasized optic nerve and spinal cord involvement, neuromyelitis optica is now believed to affect additional regions of the central nervous system, including the area postrema, brainstem, diencephalon, and cerebrum.4 The 2015 updated diagnostic criteria for neuromyelitis optica is included in Tables 1-3. When neuromyelitis optica is suspected, another demyelinating condition, myelin oligodendrocyte glycoprotein antibody disease, should be ruled out by testing for myelin oligodendrocyte glycoprotein antibody. Myelin oligodendrocyte glycoprotein antibody disease often affects younger, pediatric patient populations and more commonly presents with disc edema compared with neuromyelitis optica.1,5 Neuromyelitis optica affects middle-aged women more than men (9:1 ratio), with the average age of onset around 39 years.6 Population studies suggest a higher racial prevalence in black individuals compared to other racial groups.7 Table 4 shows additional comparisons between neuromyelitis optica, multiple sclerosis, and myelin oligodendrocyte glycoprotein antibody disease. Visual prognosis in patients with neuromyelitis optica is worse in individuals experiencing relapsing episodes, with more than half of patients experiencing permanent vision loss within 5 years of disease onset. This visual impact may include profound loss of central visual acuity, visual field defects, reduced contrast sensitivity, or color vision deficits.1,6 Visual field defects can vary in appearance but may present as diffuse, central, or altitudinal loss.8 Key predictors in successful visual recovery following a neuromyelitis optica attack is how quickly a patient receives treatment as well as their entering acuity level.9 No identifiable health information was included in this case report.
CASE REPORT
A 58-year-old Black woman presented with reports of significant visual acuity decline with a duration of 3 days. Her left eye was impacted first, but she began noticing symptoms in her right eye within 24 hours of initial symptom onset. She complained of light sensitivity in both eyes that was compared to the sensation she experiences when her eyes are pharmaceutically dilated in a clinical setting. She recently completed her routine eye examination while in physical rehabilitation at another facility for transverse myelitis 3 weeks prior. The findings of her previous optometry examination were unremarkable, with visual acuity measuring 20/20 in each eye.
The patient’s transverse myelitis was diagnosed 18 months before the onset of her ocular symptoms and caused progressive lower extremity paraplegia, paresthesia, muscle pain, and impaired bladder function. Her prior medical workup at the time of being diagnosed included positive aquaporin-4 immunoglobulin G with spinal magnetic resonance imaging showing intramedullary lesions from levels C5 to upper thoracic segments. She had no ocular involvement at the time of initial transverse myelitis diagnosis. In addition to her transverse myelitis diagnosis, her medical history was significant for hypertension that was being managed with 10 mg amlodipine and 25 mg hydralazine. She was also treated for hypercholesterolemia with 40 mg pravastatin. She did have a history of alcohol abuse but has been sober for several years.
The patient’s best-corrected visual acuity was 5/100 (20/400) in the right eye and 2/700 (20/7,000) in the left eye using a Feinbloom acuity chart. Pupillary evaluation showed a left afferent pupillary defect. She did not have any limitation in extraocular muscle function. Given her level of acuity, visual field estimation was challenging but did appear to be grossly full by confrontation without obvious neurologic defects. Her intraocular pressures were measured at 14 mmHg in the right eye and 15 mmHg in the left eye using Tono-Pen. Despite the abnormal pupillary finding, there were no other ocular abnormalities noted on anterior or posterior segment examination. Specifically, her optic nerve appeared well perfused with distinct margins and a robust neural retinal rim. Unfortunately, we were not able to obtain any additional ophthalmic imaging because of the patient’s physical limitations.
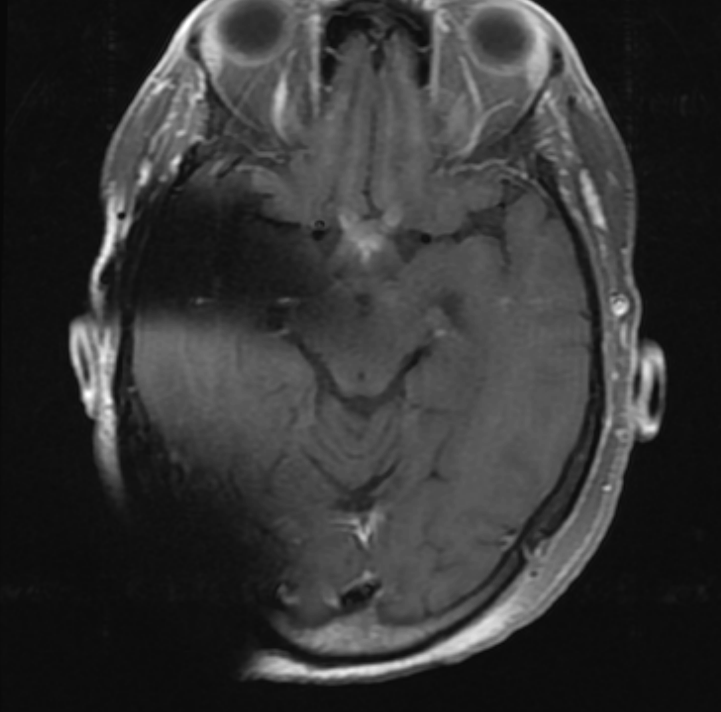
Without clear ocular etiology for profound visual decline and an afferent pupillary defect, magnetic resonance imaging with and without contrast dye was ordered of the brain. The results of this scan showed optic chiasm enhancement suggestive of bilateral, retrobulbar optic neuritis (Figure 1). There was no asymmetry in enhancement noted that would explain an afferent pupillary defect in one eye only; however, the onset of symptoms involved her left eye approximately 24 hours before her right eye which could explain this asymmetric finding. The patient’s laboratory results confirmed increased aquaporin-4 immunoglobulin G, as seen in her initial transverse myelitis diagnosis, but was negative for myelin oligodendrocyte glycoprotein antibody. These findings, in addition to the previous diagnosis of transverse myelitis, were consistent with neuromyelitis optica disorder. Additional laboratory work ruled out other autoimmune conditions, including Sjogren’s syndrome, thyroid disease, systemic lupus erythematosus, rheumatoid arthritis, and myasthenia gravis. The patient was emergently admitted to our facility to receive 1000 mg intravenous methylprednisone daily over 5 days; however, this did not initially result in improvement of her visual acuity. Her medical team then elected to continue oral prednisone beginning at 60 mg tapered over 6 weeks and to initiate plasma exchange therapy. This additional treatment ultimately was successful in improving her visual acuity after completing the 5 plasma exchange therapy sessions. After the patient’s acute phase was managed, her medical team shifted their attention to reducing the chance of relapse through the use of immunomodulating agent, rituximab. Rituximab infusion was attempted the day before the patient was discharged from the hospital, but the patient experienced a mild infusion reaction within 12 hours from treatment. The treatment plan outlined that rituximab infusions would be attempted again in an outpatient setting once the patient had time to regain her strength after her extensive, combined hospital and inpatient physical therapy stay.


Although there was subjective report of improved acuity included in her inpatient notes by her medical team, the first measurable acuity was obtained approximately 3 months later during her ophthalmology follow-up. Her acuity was measured at 20/20 in each eye. She did have some residual subjective color vision deficits in both eyes; however, color vision was still found to be normal by Ishihara. The patient continues to report some mild light sensitivity at times since her recovery. Her posterior segment examination revealed healthy appearing optic nerves without blurred margins or pallor. An automated visual field was attempted to determine if there were any consequential defects of her optic neuritis flare up; however, this test was highly unreliable and difficult to obtain because of her persistent paraplegia and paresthesia from transverse myelitis. Confrontation visual fields were performed and estimated to be full without measurable defects. This was consistent with the visual field estimations from her initial visit. Neither fundus photography nor optical coherence tomography could be performed because of her physical and positional limitations of her power wheelchair with the imaging equipment; however, these images would have been helpful in monitoring nerve appearance and detecting early changes in retinal nerve fiber layer thickness.
The patient actively receives intravenous rituximab infusions every 6 months to prevent relapsing episodes, which has shown to be successful in preventing recurrence in her case. She is tolerating these infusions better compared with initial treatment attempts, but she does continue to experience mild headaches with each infusion. Her vision remains stable, and she participates in physical therapy several times a day in an assisted-living facility to try to improve physical function. She has been discharged from ophthalmology and continues to be monitored in the optometry clinic every 6 months.
DISCUSSION
As seen in our patient, one of the hallmark features of neuromyelitis optica is that patients have acute onset optic neuritis that occurs either simultaneously or sequentially between the two eyes. Our patient also had a previous diagnosis of transverse myelitis, which may present with paresthesia, paraplegia, painful muscle spasms, bladder dysfunction, and bowel dysfunction.7 Additional autoimmune conditions, such as Sjogren syndrome and systemic lupus erythematosus, may be present in neuromyelitis optica; however, her laboratory work did not suggest any additional autoimmune diagnoses.4 The optic nerve and spinal cord are targets of neuromyelitis optica because of the shared feature of aquaporin-4 water channel, a component of the blood-brain barrier, which is targeted by aquaporin-4 immunoglobulin G; therefore, patients are at risk for both transverse myelitis and optic neuritis1,2 According to updated diagnostic guidelines, at least 3 contiguous vertebral segments must be affected for a diagnosis of transverse myelitis secondary to neuromyelitis optica.5 An area of the medulla oblongata called the area postrema can also be targeted in neuromyelitis optica. This can cause an additional symptom of intractable hiccups or vomiting that often precedes or occurs simultaneously with transverse myelitis and optic neuritis.10
The initial diagnosis of neuromyelitis optica requires careful attention to the nature of onset and the severity of symptoms. The absence of ocular findings on posterior segment examination in the presence of a relative afferent pupillary defect were highly suggestive of a retrobulbar process. An inflammatory etiology was the leading differential diagnosis given the acute onset and the patient’s medical history; however, a compressive lesion could have also been considered especially if symptom onset had been more gradual. Neuroimaging using magnetic resonance imaging with and without contrast dye can assist in ruling out both possibilities and with blood work evaluating for aquaporin-4 immunoglobulin G and myelin oligodendrocyte glycoprotein antibodies.
Visual prognosis for patients with neuromyelitis optica is heavily determined by the severity of the attack and frequency of relapsing episodes. In severe attacks, visual acuity may remain worse than 20/200 and visual field defects may be present. In addition to central and diffuse defects seen in other optic neuritis cases, neuromyelitis optica may also have altitudinal defects similar to ischemic optic neuropathy. This is thought to be related to ischemic damage from aquaporin-4 immunoglobulin G–mediated attacks.8 Generally, visual outcomes are poor compared with multiple sclerosis and myelin oligodendrocyte glycoprotein antibody disease; however, treatment of active phase within 21 days of the initial attack has been shown to have the best visual outcomes.9
The first treatment goal of neuromyelitis optica with ocular manifestations is to address the active optic neuritis using high-dose steroids. The initial recommendation is 1 gram of intravenous methylprednisone daily over 3 days and is most effective with early treatment initiation within 2 weeks of disease onset.1,11 According to the Optic Neuritis Treatment Trial, intravenous methylprednisone in combination with oral prednisone was preferred over oral prednisone in monotherapy for optimal visual recovery and in reducing relapse rate.11 Although the patient population in the Optic Neuritis Treatment Trial included those with multiple sclerosis rather than those with neuromyelitis optica, this treatment protocol was used in this case as a reference for combined intravenous and oral steroid treatment in acute optic neuritis cases.
An additional treatment goal is to reduce the risk of recurrence given the high chance for visual impairment with subsequent episodes. Up to 86% of patients have been estimated to experience relapse with 49% of patients experiencing relapse within the first year from initial onset.12 Although the efficacy of immunomodulating agents in preventing relapse continues to be studied, rituximab infusions continue to be used clinically with long-term dosing every 6 months.13 Additional approved antibody therapies include eculizumab, inebilizumab, and satralizumab.14 Of note, studies using these agents are limited because of the rarity of neuromyelitis optica and, consequently, low study patient population.
Our patient received plasma exchange therapy, which is used to separate inflammatory factors such as immunoglobulins from the plasma to reduce pathologic potential. Although there is still ongoing work to determine appropriate clinical applications, plasma exchange has primarily been used in cases that do not respond well to steroids, as was seen in this case. The success of plasma exchange in acute cases with poor steroid response suggest that plasma exchange may need to be included in first-line protocol in addition to intravenous steroids.15 As with intravenous steroids, prompt plasma exchange treatment was found to be most beneficial with early treatment, with the highest success within 11 days of neuromyelitis onset.16
CONCLUSION
Optic neuritis secondary to neuromyelitis optica poses a significant threat to vision; therefore, early recognition and treatment is imperative for successful outcomes. Treatment in these cases involve a 2-part protocol, including treating the active inflammation and reducing the risk of recurrence. The task of treating active inflammation includes a combination of intravenous steroids, oral steroids, and plasma exchange therapy, whereas immunomodulating agents have been successful in reducing relapsing episodes. This case demonstrates that clinicians can minimize the risk for additional visual impairment associated with neuromyelitis optica through timely intervention.
TAKE HOME POINTS
-
Acute, profound vision loss with the absence of ocular findings warrants emergent neuroimaging, including magnetic resonance imaging with and without contrast dye.
-
Timely initiation of intravenous steroids, in combination with an oral steroid taper in patients with optic neuritis secondary to neuromyelitis optica, is crucial to optimize the chance of successful visual recovery.
-
Plasma exchange treatment in neuromyelitis optica should be considered in cases that do not respond well to steroid treatment and may even be considered as part of first-line therapy when available.
-
Intravenous immunomodulating agents such as rituximab are beneficial for prevention of recurrence, thus reducing the risk of permanent visual impairment from neuromyelitis optica.
DISCLOSURES
The content of this case report does not represent views held by the Department of Veterans Affairs nor the United States Federal Government.